
Every protein, gene, and regulatory element is the product of billions of years of evolutionary constraint acting simultaneously on sequence, structure, and function. A sequence alone is an incomplete view of a gene or protein because its conservation profile, splice patterns, structural geometry, and chemical descriptors are all independent factors mapping onto the same underlying biological reality. Yet the AI models we use to study biology treat these dimensions as independent problems, training separate systems for DNA, RNA, and protein with no shared understanding of what they jointly encode.
This fragmentation presents fundamental limitations:
- First, these models have no mechanism to incorporate auxiliary biological information at inference time. However, a model that knows both the sequence and the evolutionary conservation profile of a transcript should theoretically make better splice predictions than one that sees sequence alone.
- Second, the inverse questions that matter most for medicine and design go unanswered: given a desired protein structure and splicing pattern, what sequence is most likely to produce them? Existing models, trained to map fixed inputs to fixed outputs, have no principled way to ask, let alone answer, that question.
We built MIMIC to address these limitations. MIMIC is a generative foundation model that jointly represents DNA, RNA, protein, and cellular context within a single unified framework, learning a shared distribution over molecular states rather than any one slice of it. The result is a model that can integrate information across the central dogma: given any observed subset of biological information, it can condition on that context to infer the remainder.
How MIMIC works
MIMIC is a billion-parameter encoder-decoder transformer trained to jointly model molecular data of the central dogma. Where most biological AI models take a single type of input and produce a single type of output, MIMIC is designed to take in any available molecular information and generate what is missing. Protein surface geometry, RNA splice patterns, evolutionary conservation scores, and natural language descriptions of experimental conditions are all first-class inputs and outputs within the same model.
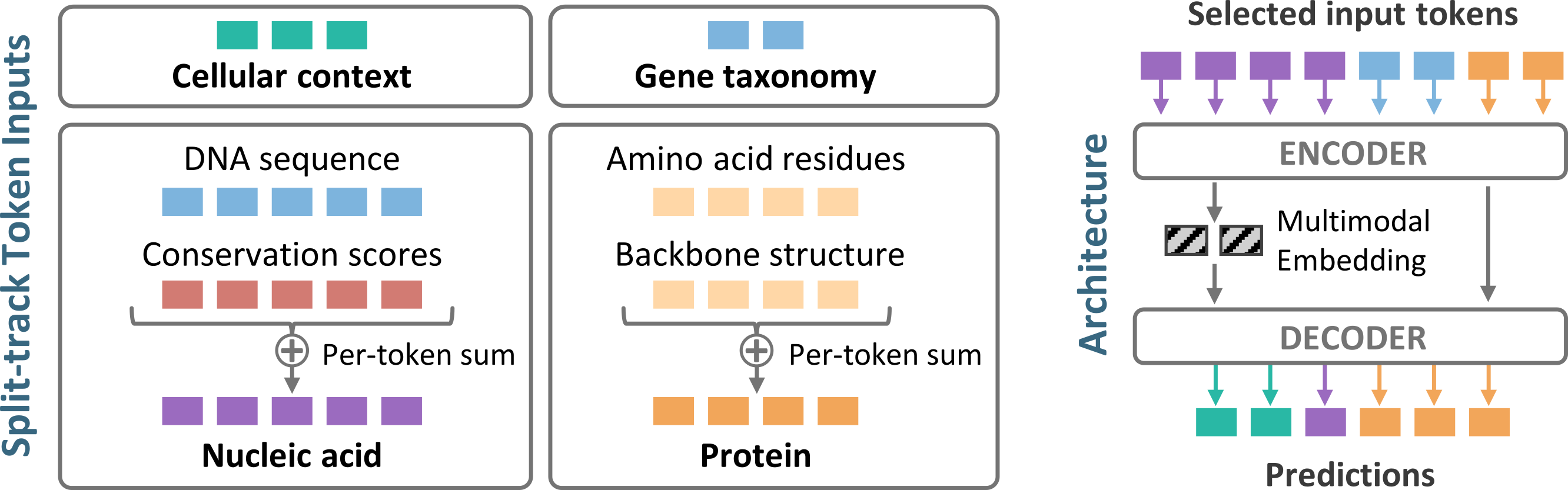
Split-track architecture. Rather than concatenating all biological signals into one long sequence, MIMIC organizes inputs into distinct track groups reflecting the natural coordinate systems of biology. Nucleotide-level signals are embedded and summed into a nucleic acid track, residue-level signals into an amino acid track, and unaligned information like free-text context into separate token groups. Adding more modalities makes each position richer without making sequences longer. A cross-attention decoder then queries the encoder’s latent representation to generate any target modality, so the same model can produce a protein sequence from surface geometry or infer a splice pattern from transcript boundaries without task-specific modifications.
Localized RoPE. Position indices reset to zero at the start of each track group, so attention patterns reflect meaningful within-track distances rather than arbitrary offsets introduced by concatenation order.
Register tokens. A small set of learnable register tokens attend to all tracks and aggregate global state across the full molecular context. They are trained via a reconstruction objective in which random token dropout forces them to encode sufficient information for the decoder to recover masked inputs, encouraging genuine compression rather than copying.
Training pathways. Because most samples contain only a subset of possible modalities, training is organized into approximately 25 pathway configurations each specifying a required and optional set of input and target modalities, ensuring rare but high-value combinations are adequately represented during training.
Curriculum learning. MIMIC is trained with a staged curriculum that progressively scales the encoder context window from 1,000 to 10,000 tokens, allowing the model to learn local sequence features before resolving long-range regulatory and structural dependencies.
State-of-the-art performance on diverse downstream benchmarks
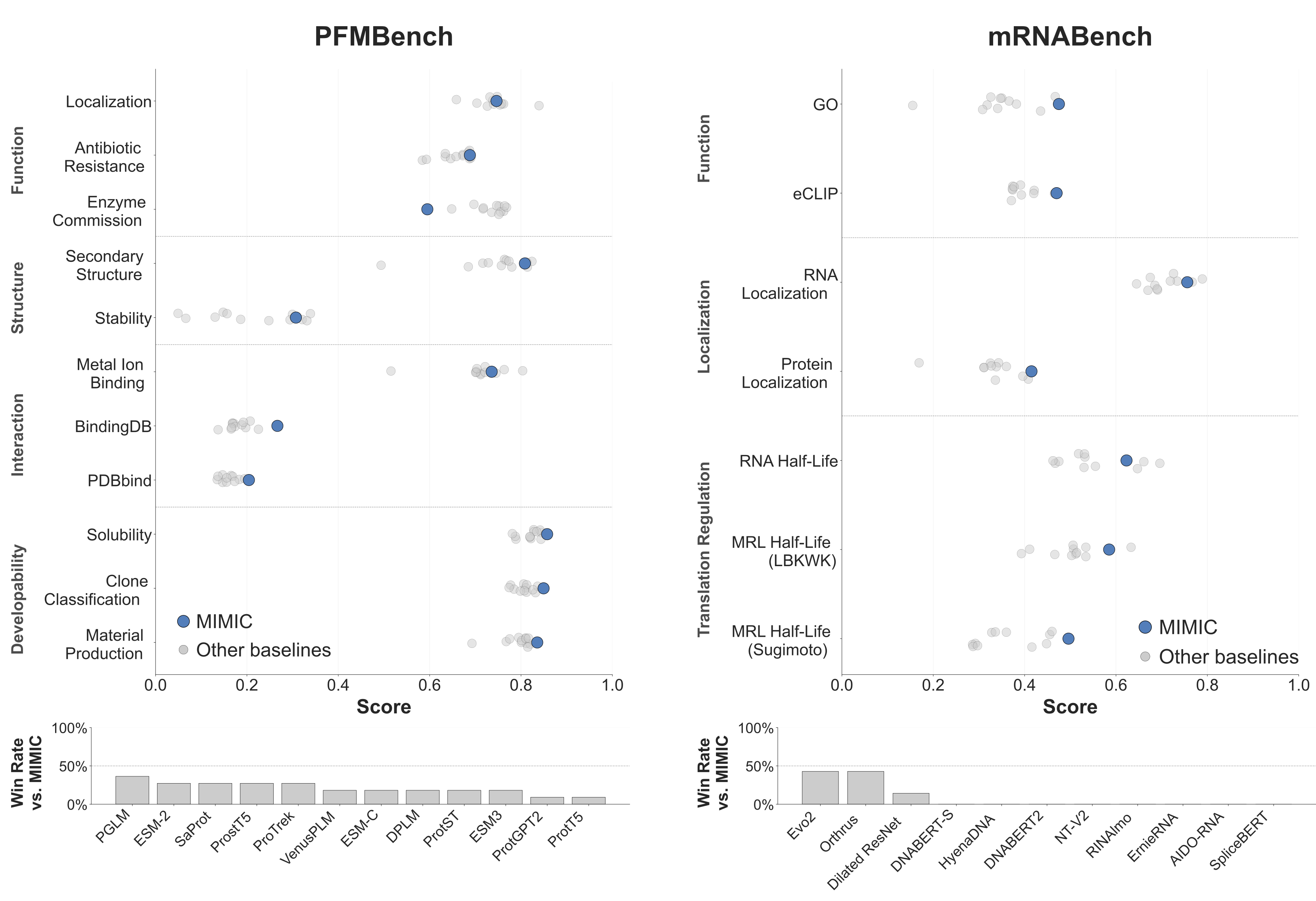
MIMIC’s representations transfer strongly across a broad range of downstream tasks. On mRNABench, covering RNA function, localization, and translation regulation, MIMIC matches or outperforms the best available public models on 6/7 tasks, with especially strong gains on GO function prediction, protein localization, and prediction of ribosome load across endogenous isoforms, suggesting that multimodal pretraining captures features tied to translational output that sequence-only models miss. On PFMBench, a comprehensive protein benchmark spanning structure, function, interaction, and developability, MIMIC ranks first or near first against strong protein-only baselines on 7/11 tasks. In both cases, performance improves further when auxiliary modalities are provided as conditioning alongside the primary sequence. The conclusion is clear: joint multimodal pretraining yields richer representations than sequence alone, even at a fraction of the parameter count.
Splicing prediction and design
Splicing is a fundamental mechanism of gene regulation, yet current models address only the forward problem: given a sequence, predict where splicing occurs. MIMIC addresses both forward prediction and inverse design. On a held-out set of human transcripts, MIMIC outperforms SpliceAI and AlphaGenome on splice site prediction. Uniquely, it can also condition on known transcript start and end sites, incorporating experimentally measured boundary information to further sharpen predictions.
The design capability is demonstrated in the setting of the well-characterized HBB IVS-II-654 C>T mutation, a deep intronic variant that disrupts normal splicing by activating cryptic splice sites and promoting pseudoexon inclusion. Conditioning on the desired wild-type splice pattern while keeping the causal mutation fixed, MIMIC redesigned nearby sequence context within editable windows of varying size and position, substantially reducing the predicted likelihood of aberrant splicing, and successful corrections were found even when the editable window did not directly overlap the cryptic splice sites. MIMIC opens up a new avenue for understanding regulatory mechanisms, where multimodal conditioning enables the model to discover indirect but biologically plausible routes to restoring regulatory function.
Multimodal-conditioned design of high-confidence protein binders
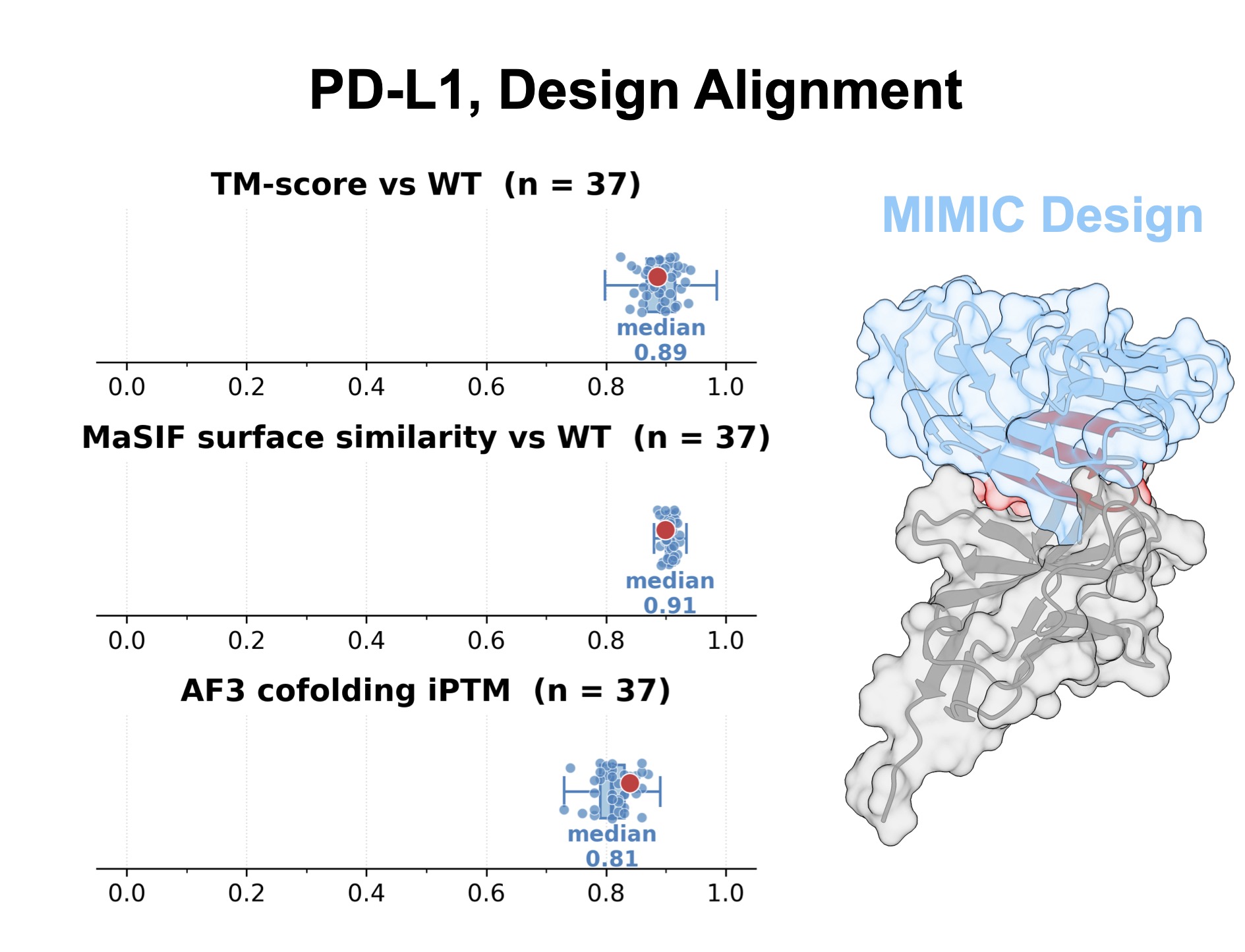
Protein design poses an analogous challenge: viable sequences must satisfy not only the geometric constraints of a target fold, but also the surface properties that govern molecular interactions. Because MIMIC jointly represents sequence, structure, and surface features, it can condition directly on both backbone geometry and the chemical environment of an interface. We tested this on two therapeutically relevant targets, PD-L1 and ACE2, finding that design quality improved as additional modalities were provided as conditioning, with joint backbone and surface conditioning producing the largest fraction of high-confidence sequences.
Designs were evaluated by predicting structures with AlphaFold2 and modeling binding with AlphaFold3; for PD-L1, this pipeline yielded a substantial fraction of high-confidence predicted binders. Crucially, these designs remained diverse in sequence space, indicating that the model explores multiple viable solutions rather than reproducing a single template. Multimodal conditioning substantially narrows the protein design search space, steering generation toward sequences that are structurally compatible and consistent with the surface properties required for binding.
Improving RNA 2D structure prediction with semantic context and predicted reactivity
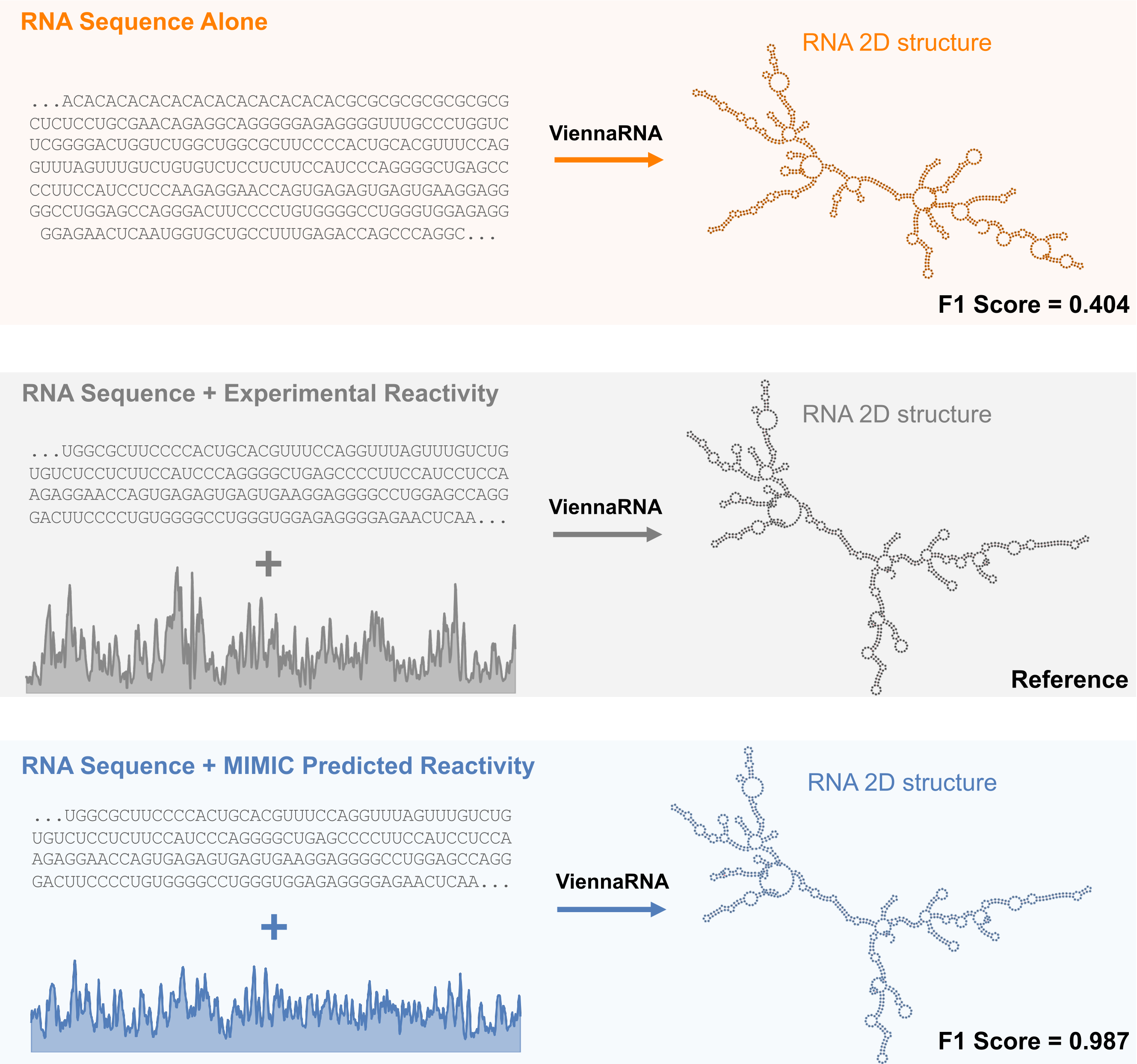
RNA secondary structure governs everything from transcript stability to translation efficiency, but experimental probing data required to resolve it accurately is available for only a small fraction of transcripts. MIMIC addresses this by conditioning on both RNA sequence and a natural language description of experimental conditions to predict chemical probing reactivity profiles (RASP2 scores). Critically, the model learns to distinguish between probing approaches and cellular environments, generating condition-specific reactivity tracks that sequence-alone models cannot produce.
These predicted profiles are directly useful. Feeding MIMIC's RASP2 predictions into ViennaRNA, a standard RNA folding algorithm, yields 2D structures that substantially outperform sequence-alone folding and are similar in accuracy to those conditioned on experimental RASP2 data. This demonstrates a concrete path from multimodal conditioning to improved structural biology: MIMIC acts as a virtual probing experiment, generating the auxiliary data needed to resolve RNA architecture at transcriptome scale.
The LORE Dataset
MIMIC’s flexibility and expressivity depends critically on LORE, a curated dataset that aligns heterogeneous biological measurements—sequence, structure, conservation, regulatory assays, surface chemistry, and semantic annotations, into coherent, partially observed molecular examples anchored to shared transcripts and proteins. Because LORE provides non-redundant, cross-modal supervision rather than isolated single-modality corpora, MIMIC can learn the complementary constraints between modalities that drive its strongest performance gains: auxiliary context helps most precisely where it provides information that sequence alone leaves ambiguous.
- Sequence
- Transcript annotation
- Splice junctions
- Coding sequence
- Evolutionary conservation
- Promoter usage
- Chromatin accessibility
- RNA chemical probing
- Sequence
- Secondary structure
- Tertiary structure
- Solvent accessibility
- Chemical surface
- Protein abundance
- Functional captions
- Taxonomic classification
- Biomedical text
- Experimental context
What comes next
MIMIC covers a meaningful subset of molecular biology, but important layers such as chromatin state, nucleic acid and post-translational chemical modifications, translational regulation, and downstream cellular phenotypes remain absent or underrepresented. Broadening modality coverage, extending context scale, and experimentally validating designed sequences are natural next steps. In its current form however, MIMIC demonstrates that joint modeling over aligned molecular state captures cross-modal dependencies that no single-modality model can access, with practical value already evident across prediction, representation learning, and generative design.
Open Source Resources
We are currently preparing MIMIC code and weights for public release. We are also preparing LORE for public release, including data clustering, splits, and pre-computed tokenization. Upon release, both the MIMIC model and the LORE dataset will be available on the Polymathic AI GitHub.
– Sophie Barstein, Samuel Sledzieski, Jake Kovalic
Acknowledgements
We would like to acknowledge the support of the Simons Foundation and of Schmidt Sciences. This work was supported in part by the AI2050 program at Schmidt Sciences (Grant G-25-70028). Additionally, computations were run at facilities supported by the Scientific Computing Core at the Flatiron Institute. The Flatiron Institute is a division of the Simons Foundation. The authors thank Justin Kinney, Doug Renfrew, and Bargeen Turzo for helpful discussions, and Lucy Reading-Ikkanda and Chi-Dat Lam for assistance with figures.
Image by Emanuel Haas via Unsplash.