The race to build AI scientists is hotter than ever, with the goal of creating AI that can perform the entire scientific process end to end. Our vision is different. Rather than building a general-purpose AI scientist, we set out to build a specialized AI agent that excels at one scientific task that can be evaluated quantitatively. Through conversations with chemists, we identified NMR structure elucidation as a recurring bottleneck accross disciplines that is time-consuming and still depends heavily on expert interpretation. This motivated us to develop an AI agent specifically for NMR structure elucidation.
The importance of NMR spectroscopy
Nuclear Magnetic Resonance (NMR) is one of the most widely used techniques for determining molecular structure. In simple terms, NMR provides a partial answer to the question of “what are the molecular structures present in this sample?” The technique leverages the known magnetic properties of atomic nuclei (the most common being hydrogen and carbon) to provide information about local chemical environments. Chemists can infer which functional groups are present in a sample and how atoms are connected by analyzing its ¹H and ¹³C NMR spectra.
NMR is a central tool across research in chemistry, biology, and materials science, as well as in industries such as pharmaceutics, food analysis, chemical manufacturing, and quality control. It is also an essential part of the discovery and assessment of the potential therapeutic value of new natural compounds that have been isolated from plants, animals, or fungi.
The acquisition of NMR data is straightforward, but its interpretation continues to be a major time bottleneck for scientists, especially in the case of complex and large structures. The process is often slow, relies on human expertise, and is difficult to automate reliably. A chemist must identify meaningful peaks, account for solvent and impurity signals, resolve overlapping regions, combine evidence across multiple spectra, and decide which molecular structures best satisfies all available constraints.
NMR Elucidation as a Molecular Puzzle
The task we are interested in is an inverse problem. NMR elucidation starts with the experimentally acquired ¹H and ¹³C NMR spectra, and the goal is to reconstruct the unknown molecular structure that gave rise to these observations. The spectra provide measurements of chemical shifts, integration values, and splitting patterns, among others, but not direct measurements of the structure. So it is up to the chemist to interpret these and guess the different local substructures of this “puzzle,” piecing together what comprises the unknown molecule and how to bond these fragments together. The spectra provide constraints that narrow the search in chemical space, but there are other verifications that chemists use.
Sometimes, the molecular formula is also available from a mass spectrometry experiment. The degree of unsaturation constrains the number of rings and multiple bonds. The ¹H NMR spectrum provides information about proton environments, relative integrations, and neighboring atoms. The ¹³C NMR spectrum provides information about carbon environments and symmetry. A correct structure must satisfy all of these pieces simultaneously.
It is also possible that multiple structures are compatible with the observed spectra, and all of the different candidates need to be evaluated by a chemist. This is why chemists sometimes call NMR elucidation a “chemical sudoku.”
Our paper builds on this iterative strategy. Rather than training a model to directly map spectra to a molecular structure, we frame NMR elucidation as constrained search. The agent proposes molecules, tests them against the molecular formula, proton count, degree of unsaturation, and expected chemical environments, then revises its hypotheses when a candidate fails to explain the evidence.
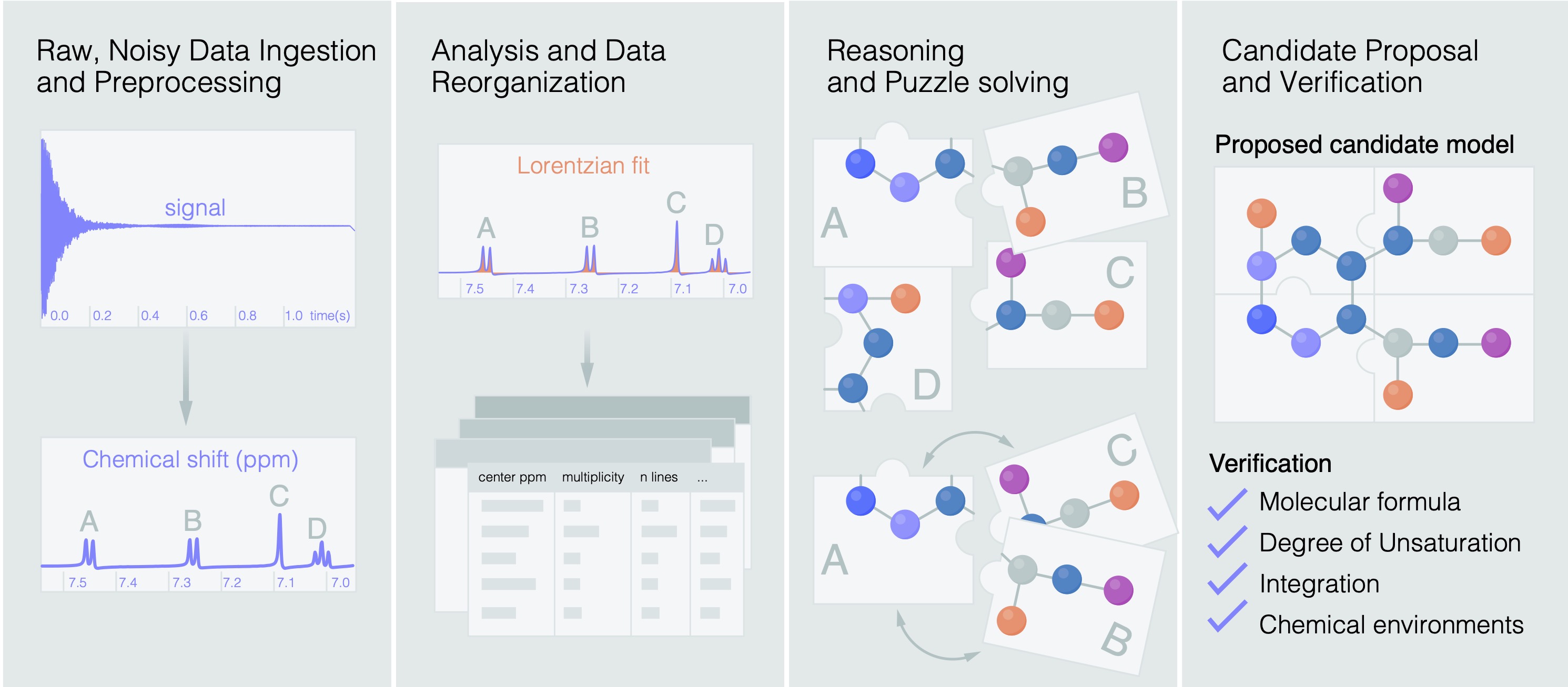
End-to-end realistic lab conditions
A key design choice in this work is that the agent starts from raw experimental NMR instrument files, rather than from hand-curated peak tables as it is common in the ML literature. A tabulated spectrum reported in a paper is already the result of human judgment because peaks have been selected, artifacts removed, integrations normalized, and ambiguous regions simplified. Such tables are useful for reporting results in a standardized way, but they do not reflect the end to end difficulty of automated structure elucidation in a laboratory setting.
Our workflow begins with raw FID files that are ingested and processed into spectra using standard NMR preprocessing steps, including Fourier transformation, phase correction, baseline correction, solvent referencing, peak picking, and integration estimation. The resulting information is organized into structured evidence tables that the agent can use to present hypothesis and re-edit.
The agent then uses these tables as a representation of the experiment. It can reason about which peaks are likely to correspond to compound molecular signals, which ones may be solvent or impurities, and which of these assignments are uncertain. This keeps as part of the task an important part of the human workflow of deciding which parts of the spectrum should be trusted. Noise, solvent peaks, impurities, overlapping signals, degraded samples, and ambiguous assignments all remain part of the input evaluation data. This makes our benchmarking more difficult than the ones based on cleaned peak lists, but it also makes it more similar to a realistic deployment.
Comparing humans, our agent, and specialized models
We evaluate our agent on three datasets of increasing difficulty: Chemistry Education (van Bramer), Alberts, and AstraZeneca, on predicting molecular structure from experimental spectra and the molecular formula. We compare against state-of-the-art ML models specialized for NMR, against human chemists (where available), and against generic coding agents used as baselines. We report top-k accuracy (%) and Tanimoto similarity to the ground-truth structure; higher is better for both. We run our agent with three different backbone models (gpt-5.4, kimi-k2.6, and qwen3.5-122b). Best result in each column is shown in bold.
Chemistry Education (van Bramer). The most accessible and clean dataset, drawn from teaching examples.
| Type | Method | Top-1 Acc. | Top-1 Tan. | Top-5 Acc. | Top-5 Tan. |
|---|---|---|---|---|---|
| Model | Alberts et al. (zero shot) | 50.3 | – | 67.1 | – |
| Model | Alberts et al. (finetuned, 5× CV) | 69.1 | – | 91.5 | – |
| Model | Alberts et al. (finetuned + 33k exp.) | 96.2 | – | 98.8 | – |
| Model | ChefNMR L (zero shot) | 56.0 | ~0.68 | ~70 | ~0.80 |
| Agent | Ours (gpt-5.4) |
80.4 | 0.88 | 88.3 | 0.93 |
| Agent | Ours (kimi-k2.6) |
80.9 | 0.90 | 90.0 | 0.93 |
| Agent | Ours (qwen3.5-122b) |
55.0 | 0.69 | 69.8 | 0.82 |
| Baseline | Codex 5.5 | 62.3 | 0.73 | 72.6 | 0.83 |
| Baseline | CC Opus-4.7 | 55.6 | 0.63 | 63.7 | 0.69 |
Alberts. A harder set of experimental spectra, with a human grad-student baseline for reference.
| Type | Method | Top-1 Acc. | Top-1 Tan. | Top-5 Acc. | Top-5 Tan. |
|---|---|---|---|---|---|
| Human | Grad. students | 66.7 | – | 84.5 | – |
| Model | Alberts et al. | 69.3 | – | 81.3 | – |
| Agent | Ours (gpt-5.4) |
66.7 | 0.76 | 71.1 | 0.80 |
| Agent | Ours (kimi-k2.6) |
71.1 | 0.80 | 77.8 | 0.82 |
| Agent | Ours (qwen3.5-122b) |
35.6 | 0.54 | 44.4 | 0.62 |
| Baseline | Codex 5.5 | 40.0 | 0.56 | 48.9 | 0.61 |
| Baseline | CC Opus-4.7 | 24.4 | 0.39 | 33.3 | 0.49 |
AstraZeneca. The most challenging dataset, drawn from real-world industrial samples.
| Type | Method | Top-1 Acc. | Top-1 Tan. | Top-5 Acc. | Top-5 Tan. |
|---|---|---|---|---|---|
| Model | Alberts et al. | 14.7 | – | 22.9 | – |
| Model | MMST (base model) | 0.0 | – | 3.0 | – |
| Model | MMST (trained on analogues) | 12.0 | – | 44.0 | – |
| Model | MMST (trained on test) | 31.0 | – | 81.0 | – |
| Agent | Ours (gpt-5.4) |
7.8 | 0.38 | 15.7 | 0.41 |
| Agent | Ours (kimi-k2.6) |
20.6 | 0.63 | 29.1 | 0.65 |
| Agent | Ours (qwen3.5-122b) |
2.0 | 0.22 | 2.9 | 0.24 |
| Baseline | Codex 5.5 | 12.7 | 0.39 | 15.7 | 0.41 |
| Baseline | CC Opus-4.7 | 6.9 | 0.26 | 7.8 | 0.28 |
A few takeaways stand out. On the easier van Bramer set, our agent is competitive with strong specialized models and clearly outperforms generic coding-agent baselines. On Alberts, our best configuration (kimi-k2.6) matches or exceeds both the human grad-student baseline and the specialized model. And on the most difficult AstraZeneca data, where every method struggles, our agent still surpasses the specialized model and coding baselines without any dataset-specific training. Across all three benchmarks, the choice of backbone model matters a great deal.
Failure modes
The agent is far from solving NMR elucidation and automating the task, looking at where it breaks down is instructive to keep improving. As molecules and their spectra become more complex, the task gets harder. We can see this by stratifying performance on the Chemistry Education dataset along two axes of complexity: the heavy-atom count of the molecule (a proxy for molecular size) and the number of unique ¹³C environments (a proxy for spectral complexity). In both cases, accuracy tends to fall as complexity rises.
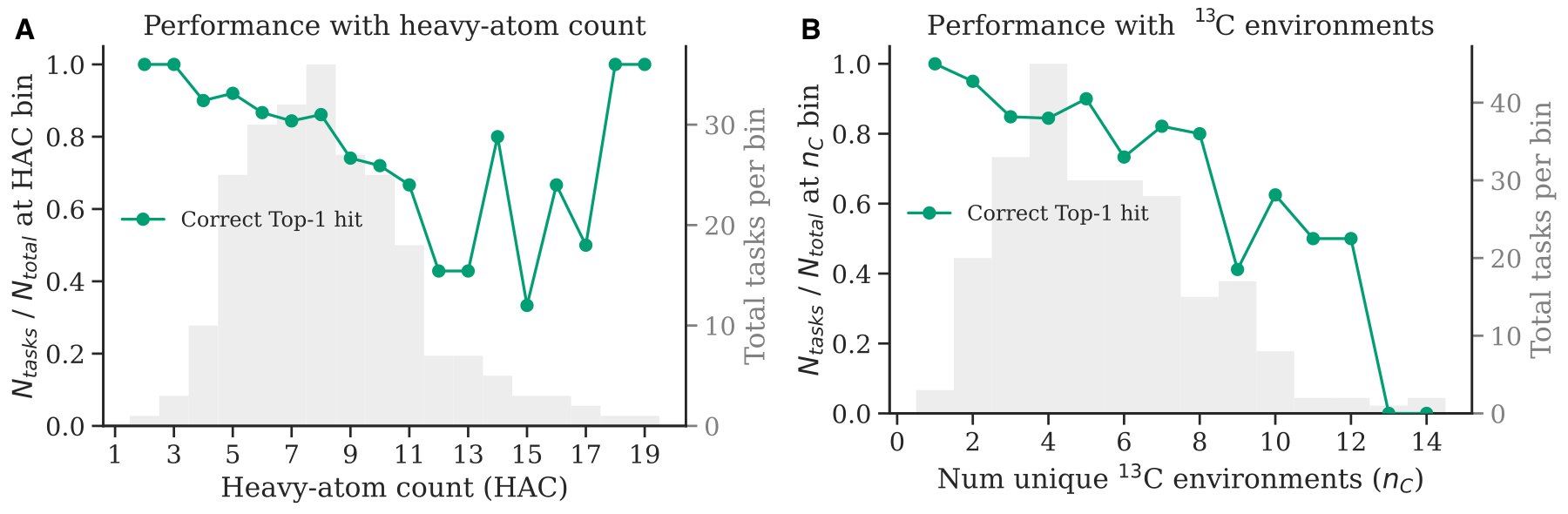
Looking at the individual predictions, three failure modes stand out:
- Near-misses: the predicted structure is very close to the ground truth (high Tanimoto similarity) but differs in one connectivity detail.
- Structural failures: the agent commits early to the wrong molecular family and cannot escape it, sometimes entering a long reasoning loop that derails.
- Stereochemical errors: the connectivity is correct, but the predicted stereochemistry differs — which we count as a miss.
Reading the agent’s reasoning traces makes these concrete. In the following (heavily condensed) trace, the agent anchors on an incorrect oxazoline/benzimidazole family and keeps explaining away each piece of disconfirming evidence and even questioning the trusted molecular formula, rather than abandoning the hypothesis:
A subtler mode is the near-miss, where the correct answer is actually within reach. On fenbufen, the agent recovered every fragment correctly and even placed the right isomer in its own candidate list, but the single diagnostic clue (a 174 ppm carbon is an aliphatic acid at ~174–180 ppm, not an aromatic one at ~167–172 ppm) was lumped into a coarse 170–175 ppm range, so it never used it to rule out its own benzoic-acid answer:
These traces point to a common thread: the agent’s biggest weakness is not raw chemical knowledge but revising an early commitment in the face of ambiguous or degraded evidence.
Looking ahead
As the failures above show, the current system is not a complete solution to NMR structure elucidation, nor a replacement for chemists. Performance decreases as molecules become larger, spectra become noisier, and the number of possible assignments grows. The framework naturally extends beyond ¹H and ¹³C NMR. In practice, chemists often combine NMR with IR, mass spectrometry, 2D NMR, Raman, UV-vis, EPR, and other modalities. A future multi-agent system could assign specialized roles to preprocessing, spectral interpretation, candidate generation, verification, and cross-modal reasoning.
For more details, check out the paper.
Acknowledgements. We thank Lucy Reading-Ikkanda at the Simons Foundation for her help with graphic design. We would like to acknowledge the support of the Simons Foundation and of Schmidt Sciences. This work was supported in part by the AI2050 program at Schmidt Sciences (Grant G-25-70028). We thank the Flatiron Institute’s Scientific Computing Core for ongoing support. The computations reported in this paper were performed using resources made available by the Flatiron Institute. The Flatiron Institute is a division of the Simons Foundation.